EPIGENETIC ANALYSIS OF PHOT-1
Epigenetics, literally meaning "on top of" the genes, refers to the modifications that are made to DNA that affect its behavior. Some of these changes are caused by environmental factors, and some are inheritable. This implies that a certain amount of Lamarckian inheritance is possible. A major way DNA can be modified is through methylation.
Methylation
DNA methylation, as shown in Figure 1, occurs when certain cytosine residues have a methyl group added, becoming 5-methylcytosine. Although methylation can have different effects, the most well-studied effect is that methylation turns off genes. This is especially important in multicellular organisms, because certain cell types have no need for certain genes. For example, a pancreatic cell has no use for the crystallins found in the lens, and any of this ectopic protein is a wasted effort. Methylation helps to completely silence a gene.
Transposable elements are also very often silenced by methylation. These so-called "jumping-genes" are very common throughout eukaryotic genomes. If they are not turned off, they may insert themselves in the middle of genes, destroying their function. In fact, mutant strains that cannot methylate these transposons often become nonviable after a few generations, as transposons wreak havoc on the genome.
Researchers can study methylation by chemically converting cytosine to uracil; 5-methylcytosine is protected from this conversion. Comparing control and converted DNA allows one to see the locations of methylated cytosine.
In Arabidopsis, there are three mechanisms of methylation that have been identified.
1. MET1, a homolog to the mammalian DNMT1, maintains methylation at CG sites.
2. CMT3 maintains methylation at CHG sites, where H is A, C, or T.
3. DRM1 and DRM2 maintain methylation at CHH sites using small RNAs (smRNAs).
Transposable elements are also very often silenced by methylation. These so-called "jumping-genes" are very common throughout eukaryotic genomes. If they are not turned off, they may insert themselves in the middle of genes, destroying their function. In fact, mutant strains that cannot methylate these transposons often become nonviable after a few generations, as transposons wreak havoc on the genome.
Researchers can study methylation by chemically converting cytosine to uracil; 5-methylcytosine is protected from this conversion. Comparing control and converted DNA allows one to see the locations of methylated cytosine.
In Arabidopsis, there are three mechanisms of methylation that have been identified.
1. MET1, a homolog to the mammalian DNMT1, maintains methylation at CG sites.
2. CMT3 maintains methylation at CHG sites, where H is A, C, or T.
3. DRM1 and DRM2 maintain methylation at CHH sites using small RNAs (smRNAs).
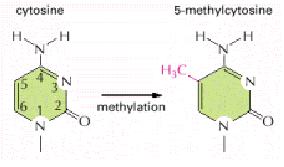
Figure 1. DNA methylation. From Molecular Biology of the Cell 4th Edition.
Epigenome browser
The SALK institute maintains a tool called the Epigenome Browser, available here. Like the TAIR G-Browser, this tool can display information from many different fields of study. It contains 5-methylcytosine mapping data for the whole Arabidopsis thaliana genome at single-base resolution. In addition, it can show data for mRNA and siRNA transcription levels, as based on the number of reads observed for a certain area.
There are four different genotypes represented in the browser:
Col-0: This represents wild-type Arabidopsis thaliana.
met1: This is a line of A. thaliana that possesses a faulty Met1 gene, meaning that it cannot methylate at CG sites.
dcc: This is a triple-mutant line that affects de-novo methylation at CHH sites.
rdd: This is a triple-mutant line that affects demethylase activity, making methylation accumulate.
Figure 2 below shows the Epigenome Browser zoomed in on Phot-1 (At3g45780), comparing Col-0 (Wild type) and met1 (deactivated Met1). The DNA was collected from immature floral tissue.
There are four different genotypes represented in the browser:
Col-0: This represents wild-type Arabidopsis thaliana.
met1: This is a line of A. thaliana that possesses a faulty Met1 gene, meaning that it cannot methylate at CG sites.
dcc: This is a triple-mutant line that affects de-novo methylation at CHH sites.
rdd: This is a triple-mutant line that affects demethylase activity, making methylation accumulate.
Figure 2 below shows the Epigenome Browser zoomed in on Phot-1 (At3g45780), comparing Col-0 (Wild type) and met1 (deactivated Met1). The DNA was collected from immature floral tissue.
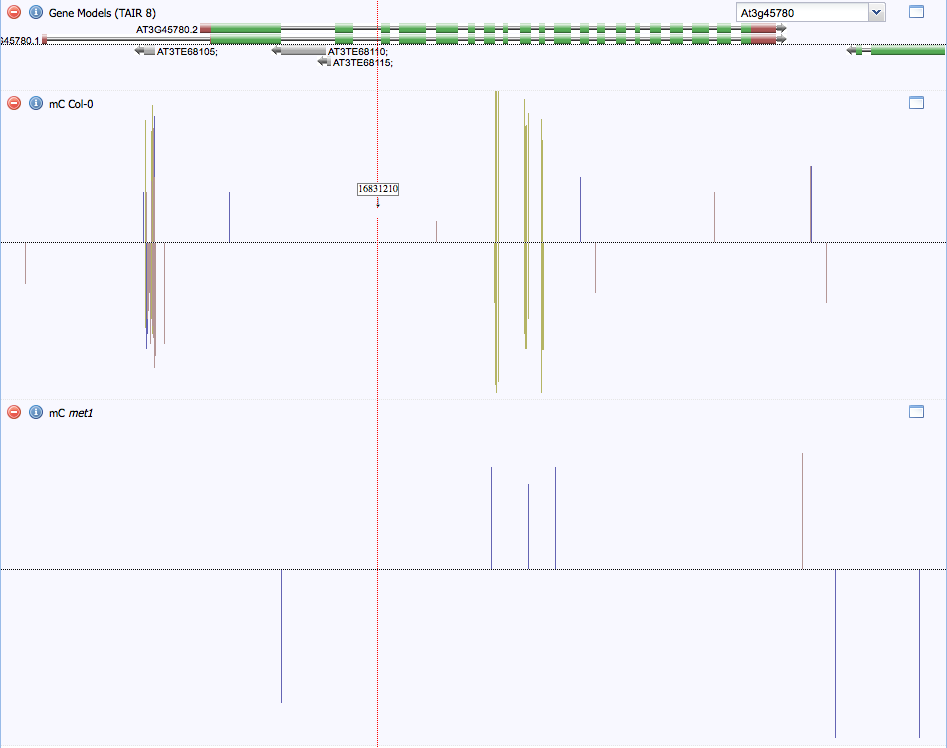
Figure 2. Methylation patterns around At3g45780 for Col-0 (wild-type) and met1 (deactivated Met1) plant lines. The protein model is shown at the top, with gray models showing predicted transposable elements. Yellow lines represent CG methylation sites. CG methylation is absent in the met1 strain. The methylated area in the first intron may be due to a small transposable element.
Figure 2 above shows that there is a small amount of 5-methylcysteine located within the Phot-1 gene. There is a significant amount of methylation in the first intron, which suggests that there may be a transposable element here that is silenced. There is a small amount of methylation in the middle of the gene, as well. Most of the methylation is done by Met1, which performs mostly maintenance methylation.
Although there is some methylation within Phot-1, there is much more that occurs in neighboring transposable elements, as seen in Figure 3. To the left of Phot-1 is AT3TE68090, also known as At3g45775, which is most definitely a transposable element. It is highly methylated at CG sites by Met1 and at CHG sites. Also highly methylated is At3g45800, a putative protein of "unknown function." This may also be a transposable element, but it might also be a protein that is not needed in the immature floral tissue and therefore silenced.
Although there is some methylation within Phot-1, there is much more that occurs in neighboring transposable elements, as seen in Figure 3. To the left of Phot-1 is AT3TE68090, also known as At3g45775, which is most definitely a transposable element. It is highly methylated at CG sites by Met1 and at CHG sites. Also highly methylated is At3g45800, a putative protein of "unknown function." This may also be a transposable element, but it might also be a protein that is not needed in the immature floral tissue and therefore silenced.
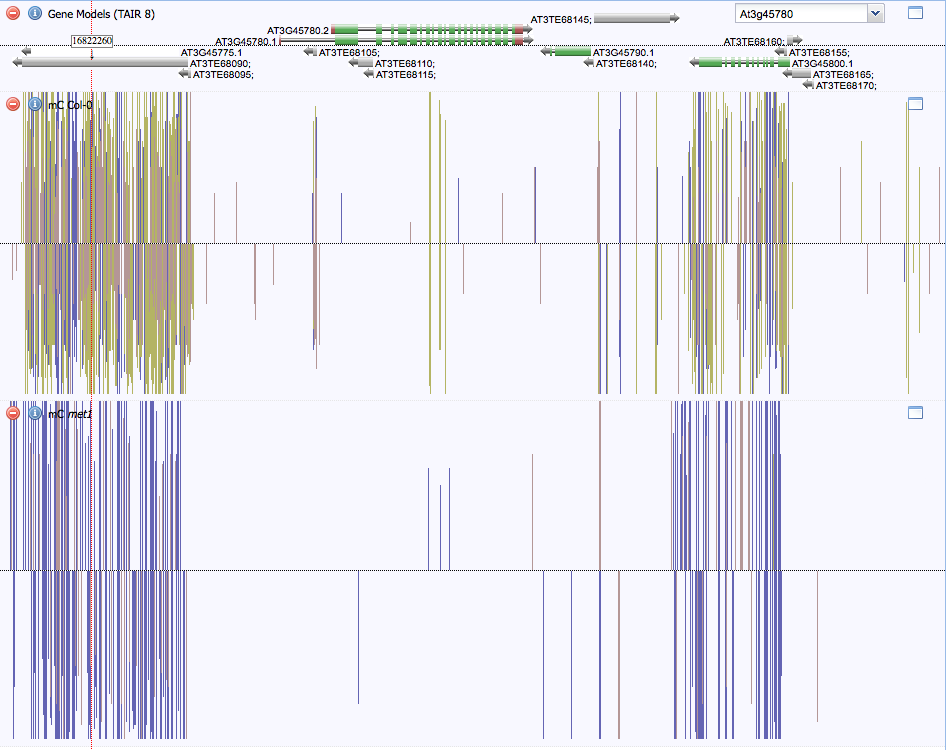
Figure 3. Methylation neighborhood. Phot-1 (At3g45780) is shown in the middle. To the left is AT3TE68090 (also known as At3g45775), which shows a high degree of methylation both in wild-type and met1 strains. It is probably a transposable element that is silenced to prevent genome damage. To the right is At3g45800, a gene of unknown function, that is also highly methylated. This gene may be methylated in immature floral tissue, the source of the DNA, but unmethylated in other tissues.
small-RNA association
The methylation mediated by DRM1 and 2 is mediated by small-RNAs, or smRNAs. Such small RNA molecules have recently been discovered to be biologically relevant. RNA interference (RNAi) is a mechanism in which smRNAs guide enzymes that destroy matching mRNAs and inhibit their translation. Similarly, smRNAs can guide methylases to certain genes, like destructive transposons, so that they can be methylated and thereby silenced.
Though not shown, the neighboring transposable element At3TE68090, or At3g45775, is associated by a large number of smRNAs, which act to silence it through smRNA-mediated methylation.
As shown in Figure 4 below, the only place in the Phot-1 gene that has any associated smRNAs is the purported transposable element At3TE68105 located in the first intron. Interestingly, no smRNAs were found to be associated to the mutant strain Met1. This may suggest that methylation at CG sites by Met1 causes enhanced smRNA association. With these kinds of experiments, however, a negative result is not very informative, because there are many technical reasons as to why smRNA may not be detected.
Though not shown, the neighboring transposable element At3TE68090, or At3g45775, is associated by a large number of smRNAs, which act to silence it through smRNA-mediated methylation.
As shown in Figure 4 below, the only place in the Phot-1 gene that has any associated smRNAs is the purported transposable element At3TE68105 located in the first intron. Interestingly, no smRNAs were found to be associated to the mutant strain Met1. This may suggest that methylation at CG sites by Met1 causes enhanced smRNA association. With these kinds of experiments, however, a negative result is not very informative, because there are many technical reasons as to why smRNA may not be detected.
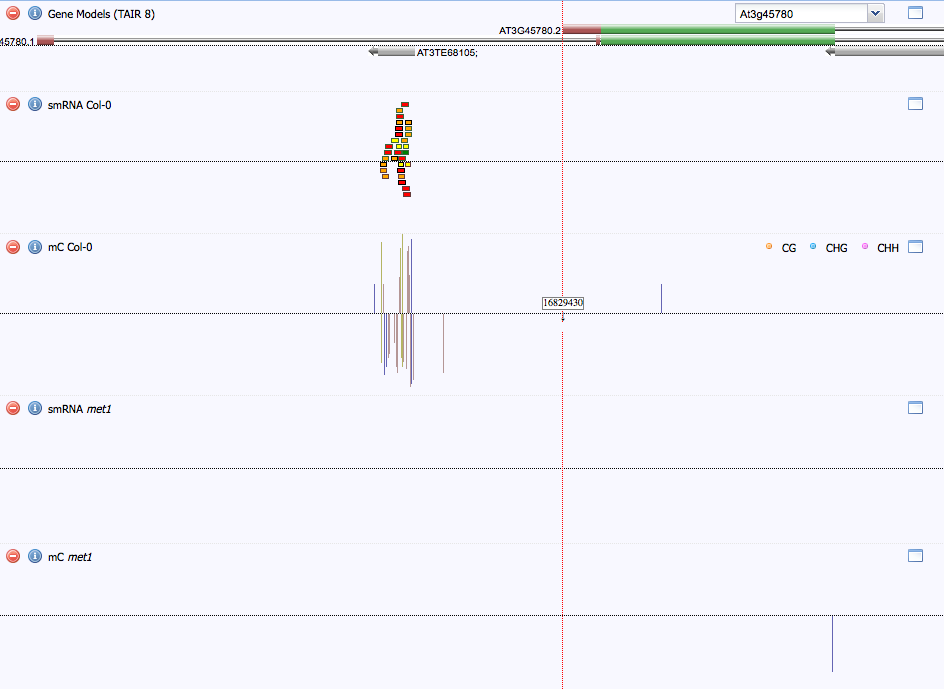
Figure 4. smRNA association. smRNA association was only observed at a putative transposable element (At3TE68105) in the first intron of Phot-1. This association appears to be abolished in the met1 mutant strain.
Estimating Transcription levels
High-throughput mRNA sequencing allows for a crude way to measure transcription levels. The sequencing technology works by sequencing very small (c. 20 bases) segments of the genome and then fitting them together by aligning overlapping segments. The number of reads of a particular segment of the genome is roughly correlated with the amount of gene expression. As an extreme example, if 75% of a cell's protein is produced by a single gene X, then that gene will have to make a large amount of corresponding mRNA. If a researcher sequences the mRNA in this cell, he or she will come across a large number of sequencing reads originating from gene X.
Figure 5 below shows the number of sequencing reads within the mRNA produced by the Phot-1 gene. No reads come from the introns because these are spliced out when mRNA is processed. Evidently, the sequencing reads of some exons are more common than others. This is most likely an artifact of the sequencing technology, because each mRNA transcript should contain every exon of the gene, barring alternative splicing.
When comparing between Col-0 (wild-type) and the Met1 strain, there is little gross difference between transcription levels, as estimated by these sequencing reads. This suggests that CG methylation by Met1 has little effect on Phot-1 transcription levels in immature floral tissue. This is probably because the gene lacks significant methylation; it is already turned on in this tissue.
Figure 5 below shows the number of sequencing reads within the mRNA produced by the Phot-1 gene. No reads come from the introns because these are spliced out when mRNA is processed. Evidently, the sequencing reads of some exons are more common than others. This is most likely an artifact of the sequencing technology, because each mRNA transcript should contain every exon of the gene, barring alternative splicing.
When comparing between Col-0 (wild-type) and the Met1 strain, there is little gross difference between transcription levels, as estimated by these sequencing reads. This suggests that CG methylation by Met1 has little effect on Phot-1 transcription levels in immature floral tissue. This is probably because the gene lacks significant methylation; it is already turned on in this tissue.
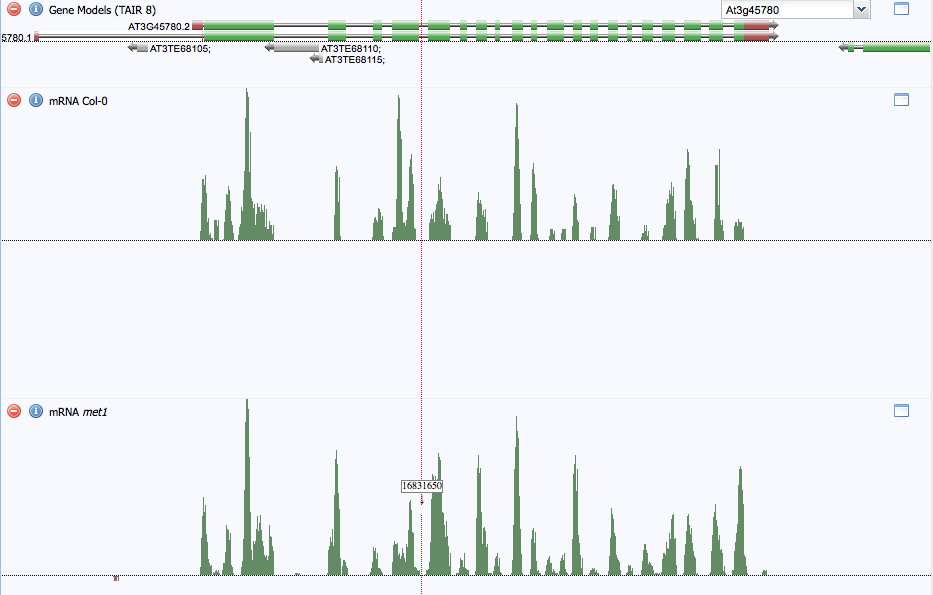
Figure 5. Sequence read comparison for mRNA between Col-0 (wild-type) and met1 mutant strains. There are no sequence reads within introns, because they are spliced out in mRNA. Sequence read levels differ between exons. Transcription levels do not seem to vary significantly between Col-0 (wild-type) and the met1 mutant strain.
summary
Epigenetics is turning out to be an important factor in the pathway from genotype to phenotype. One type of epigenetic modification is methylation, which turns cysteine into 5-methylcysteine. One effect of this modification is that it can turn the gene off. Such gene silencing is often seen in transposable elements, which would otherwise cause damage to the genome through their random insertions. The SALK institute's Epigenetics Browser allows researchers to view such methylation, at single-base resolution, throughout the Arabidopsis genome. By showing the methylation in mutant strains, one can determine which kind of methylation is most important for a given section of the genome. Additionally, the Browser can show small RNA association, which can act to guide certain enzymes to a gene in order to methylate it. Viewing sequencing read numbers allows for a rudimentary measure of gene expression for individual genes.
There is little methylation in Phot-1 in the tissues used for this experiment. However, this may not be the case for other tissues, which may turn off Phot-1 if it is not being used. In certain segments of the root, for example, which may not need high expressions of Phot-1, these cells may contain methylated Phot-1 genes. Methylation could be measured for various tissues to determine if methylation has any role to play in regulating Phot-1 transcription throughout the plant.
There is little methylation in Phot-1 in the tissues used for this experiment. However, this may not be the case for other tissues, which may turn off Phot-1 if it is not being used. In certain segments of the root, for example, which may not need high expressions of Phot-1, these cells may contain methylated Phot-1 genes. Methylation could be measured for various tissues to determine if methylation has any role to play in regulating Phot-1 transcription throughout the plant.
