variation in natural Arabidopsis populations
All of the bioinformatics data explored thus far has been focused on gathering as much data as possible for a very limited number of Arabidopsis thaliana strains. Bioinformatics technology has advanced to the point that it is becoming possible to gather equally as much data, but for multiple strains. The 1001 genomes project (modeled after the human-based 1000 genomes project) aims to sequence the entire genome of 1,001 different strains of A. thaliana, gathered from sites around the world. This will allow researchers to study the natural variation present amongst A. thaliana populations.
1,001 genomes
Although the goal of 1,001 A. thaliana genomes is a long way off, there has been significant work done already. In 2010, Cao et al. released their data showing single nucleotide polymorphisms (SNPs), structural variations (SV), and highly divergent regions from their pool of 80 A. thaliana genomes. Figure 1 below shows G-Browser pointed at Phot-1 (At3g45780) showing structural variation deletions (SV deletions). Barring one exception, all of these deletions occur within introns. This suggests that each exon is important enough that a large deletion inside an exon ruins the function of the protein as a whole, or at least is detrimental enough to be bred out by natural selection.
The large amount of SVs between exons 13 and 14, shown in Figure 1 above, is of note. Perhaps there is some advantage in deleting part of this intron. This intron is also a highly divergent region, as shown in Figure 2 below.
Figure 3 below shows a third kind of variation, single-nucleotide polymorphisms, or SNPs. If these occur on a the third base of a codon (the "wobble" base), it will be a "silent" mutation that does not affect the protein's function. Therefore, SNPs can occur inside essential exons, but with the wobble restriction. They can occur with greater ease within introns, which can be modified more significantly without affecting protein function. Interestingly, there is a high SNP frequency within the fifth exon of At3g45780.1.
Figure 4 shows the variation neighborhood of Phot-1. Areas between genes are especially susceptible to SV deletions. Perhaps due to a high amount of Highly Diverged Regions in these areas, SNP frequency is low here. Neighboring gene At3g45775, a transposable element, has an unusually high amount of Highly Divergent Regions and SNPs. A probable explanation for such accumulation is that these genes, as shown in the Epigenetics page, are silenced through methylation. Because they are turned off, they may accumulate changes without any significant effect to the organism's survival.
The large amount of SVs between exons 13 and 14, shown in Figure 1 above, is of note. Perhaps there is some advantage in deleting part of this intron. This intron is also a highly divergent region, as shown in Figure 2 below.
Figure 3 below shows a third kind of variation, single-nucleotide polymorphisms, or SNPs. If these occur on a the third base of a codon (the "wobble" base), it will be a "silent" mutation that does not affect the protein's function. Therefore, SNPs can occur inside essential exons, but with the wobble restriction. They can occur with greater ease within introns, which can be modified more significantly without affecting protein function. Interestingly, there is a high SNP frequency within the fifth exon of At3g45780.1.
Figure 4 shows the variation neighborhood of Phot-1. Areas between genes are especially susceptible to SV deletions. Perhaps due to a high amount of Highly Diverged Regions in these areas, SNP frequency is low here. Neighboring gene At3g45775, a transposable element, has an unusually high amount of Highly Divergent Regions and SNPs. A probable explanation for such accumulation is that these genes, as shown in the Epigenetics page, are silenced through methylation. Because they are turned off, they may accumulate changes without any significant effect to the organism's survival.
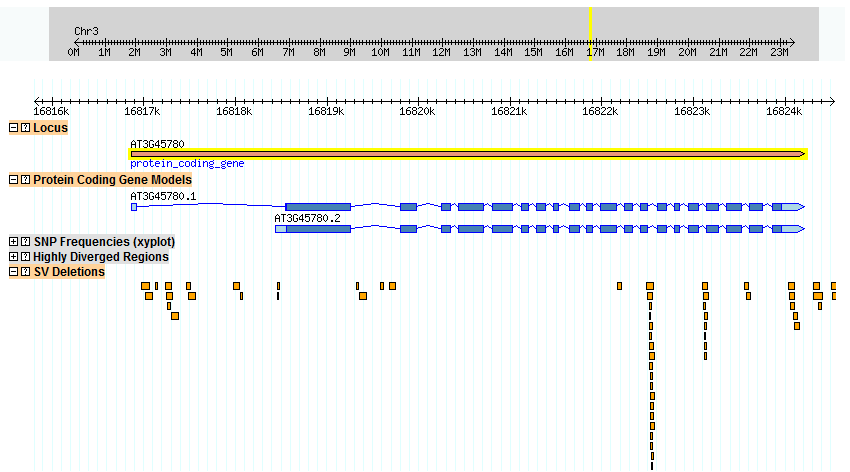
Figure 1. Structural Variations Deletions (SVDs) within the Phot-1 (At3g45780) gene. Except for a small overlap for one into exon 13 by a single ecotype, all SVDs occur within introns. The intron between exons 13 and 14 is particularly prone to SVDs. Because introns are spliced out, it is unlikely that these will significantly affect protein function.

Figure 2. Highly divergent regions within the Phot-1 (At3g45780) gene. These regions do not occur within any Phot-1 exon. They are concentrated in the first and second introns, which are the longest. The intron between exons 13 and 14 is also, unexpectedly, a highly divergent region.
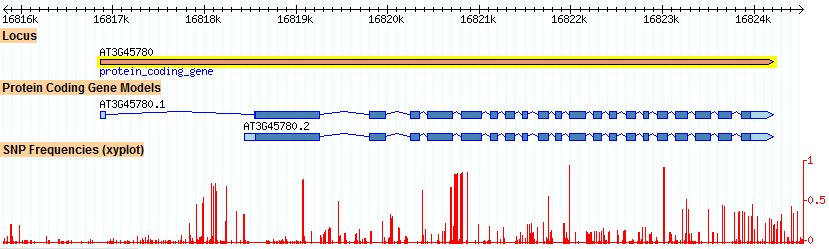
Figure 3. Single nucleotide polymorphism (SNP) frequencies in the Phot-1 (At3g45780) gene. The highest amount of polymorphisms occurs within the first intron, with a smattering elsewhere. There is a high frequency of SNPs in the fifth intron.
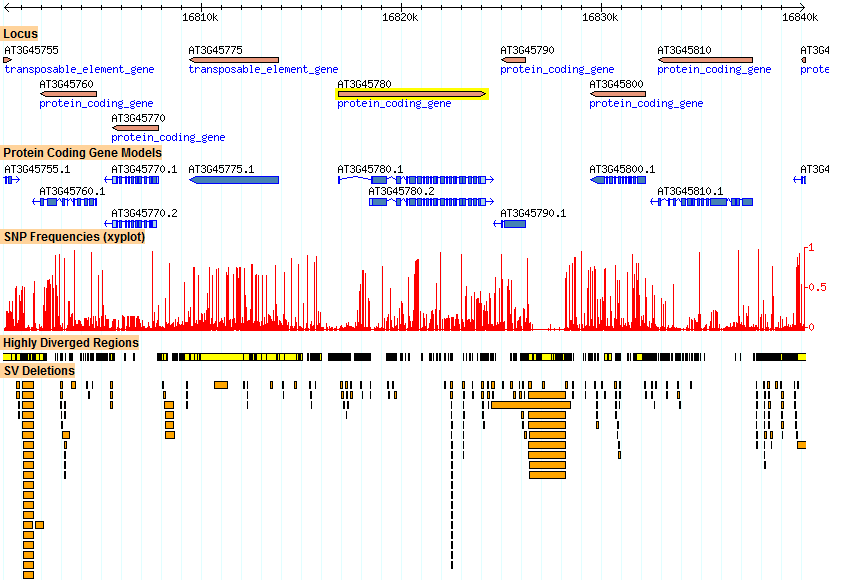
Figure 4. A zoomed-out view of variation, centered around Phot-1 (At3g45780). Areas between genes are especially susceptible to SV deletions, shown in orange at the bottom. Perhaps due to a high amount of Highly Diverged Regions in these areas, SNP frequency is shown to be low here. Neighboring gene At3g45775, a transposable element, has an unusually high amount of Highly Divergent Regions and SNPs.
1,001 proteomes
As described on the BLAST page, it is possible to predict amino acid sequences from the nucleic acid sequence. Using the nucleic acid information from the 1,001 genomes project, it is trivial to predict which SNPs produce amino acid changes for each protein. This is done using the 1,001 proteomes tool. Using the tool to view Phot-1, there are 20 SNPs that have been observed to occur in at least one ecotype that changes the amino acid.
Comparing Phot-1's amino acid sequence to distantly related plants through BLAST can reveal which sections of the gene are the most highly conserved. See a comparison to Pisum sativum, or the common pea, in this text file. Amino acid residues numbered 680 to about 770 are fairly well conserved, even between these distant relatives. This suggests that the ecotypes containing the amino-acid-changing SNPs at positions 694 and 743 may have distinct phenotypes in regards to Phot-1 and, if their seeds can be found, would be interesting to study.
Comparing Phot-1's amino acid sequence to distantly related plants through BLAST can reveal which sections of the gene are the most highly conserved. See a comparison to Pisum sativum, or the common pea, in this text file. Amino acid residues numbered 680 to about 770 are fairly well conserved, even between these distant relatives. This suggests that the ecotypes containing the amino-acid-changing SNPs at positions 694 and 743 may have distinct phenotypes in regards to Phot-1 and, if their seeds can be found, would be interesting to study.
| blastp_pisum_sativum.txt |
summary
As bioinformatics technology continues to advance, the cost of sequencing a genome continues to drop. It has become feasible to sequence entire genomes of hundreds of individual organisms, such as A. thaliana. The purpose of such an endeavor is to reveal the amount of natural variation present between naturally-occurring strains of A. thaliana gathered from different places from around the world. Although the project is not yet complete, a significant amount of data has been gathered already. Using G-Browse and a small subset of this data, the natural variation present in Phot-1 can be seen. Phot-1 is basically immune to significant changes in exons; virtually all structural variation deletions are contained within introns. The research has also unearthed a surprising amount of variation in the intron between exons 13 and 14. Further research is needed to explain this phenomenon.
By translating nucleic acid sequences to amino acid sequences, it is possible to see which SNPs cause actual amino acid changes. Of a small subset of A. thaliana genomes, 20 of these SNPs are observable on the 1,001 proteomes tool. A couple of these occur in highly-conserved regions of the gene. Studying these ecotypes in relation to Phot-1 function may reveal further insight into the gene and protein's function.
By translating nucleic acid sequences to amino acid sequences, it is possible to see which SNPs cause actual amino acid changes. Of a small subset of A. thaliana genomes, 20 of these SNPs are observable on the 1,001 proteomes tool. A couple of these occur in highly-conserved regions of the gene. Studying these ecotypes in relation to Phot-1 function may reveal further insight into the gene and protein's function.
